|
Kosloff
Lab
home
papers
people
haifa
non-science
contact
|
|
|
|
Papers (circa 2019)
|
|
 |
|
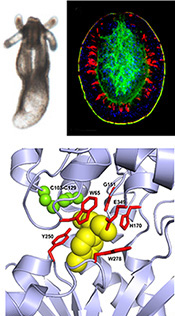
Signaling via GABA-B receptors regulates early development and neurogenesis in the basal metazoan Nematostella vectensis.
Shani Levy, Vera Brekhman, Anna Bakhman, Arnau Sebe-Pedros, Mickey Kosloff and Tamar Lotan.
Under Review (2019)
bioRxiv
The metabotropic gamma-amino-butyric acid B receptor (GABAbR) is a G protein–coupled receptor that mediates neuronal inhibition by the neurotransmitter GABA. Here, we identified putative GABAb receptors and signaling modulators in the basal sea anemone Nematostella vectensis. Activation of GABAbR signaling reversibly arrests planula-to-polyp transformation during early development and affects the neurogenic program. We identified four Nematostella GABAbR homologs that have the conserved 3D extracellular domains and residues needed for binding of GABA and the GABAbR agonist baclofen. Transcriptomic analysis, combined with spatial analysis of baclofen-treated planulae, revealed that baclofen down-regulated pro-neural factors such as NvSoxB(2), NvNeuroD1 and NvElav1. Baclofen also inhibited neuron development and extended neurites, resulting in an under-developed and less organized nervous system. Our results shed light on cnidarian development and suggest an evolutionarily conserved function for GABAbR in regulation of neurogenesis, highlighting Nematostella as a new model system to study GABAbR signaling.
|
|
 |
|
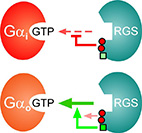
RGS6 and RGS7 discriminate between the highly-similar Gαi and Gαo
proteins using a two-tiered specificity strategy.
Ran Israeli, Ali Asli, Meirav Avital-Shacham and Mickey Kosloff
J. Mol. Biol. (2019).
web
RGS6 and RGS7 are RGS proteins that mediate diverse biological functions such as cardiac and neuronal signaling.
Uniquely, both RGS6 and RGS7 can discriminate between Gαo and Gαi -
two similar Gα subunits that belong to the same Gi sub-family.
Here, we show that the isolated RGS domains of RGS6 and RGS7 are sufficient to achieve this specificity. We identified three specific RGS6/7 "disruptor residues" that can attenuate RGS interactions towards Gα subunits and demonstrated that their insertion into a representative high-activity RGS causes a significant, yet non-specific, reduction in activity. We further identified a unique "modulatory" residue that bypasses this negative effect, specifically towards Gαo. Hence, the exquisite specificity of RGS6 and RGS7 towards closely-related Gα subunits is achieved via a two-tier specificity system, whereby a Gα-specific modulatory motif overrides the inhibitory effect of non-specific disruptor residues. Our findings expand the understanding of the molecular toolkit used by the RGS family to achieve specific interactions with selected Gα subunits - emphasizing the functional importance of the RGS domain in determining the activity and selectivity of RGS R7 sub-family members towards particular Gα subunits.
|
|
 |
|
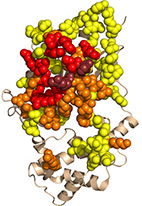
Structural design principles that underlie the multi-specific interactions of Gαq with
dissimilar partners.
Shir Navot and Mickey Kosloff
Sci. Rep. (2019) 9 (1): 6898.
web
pdf
Gαq is a ubiquitous molecular switch that activates the effectors phospholipase-C-β3 (PLC-β3) and Rho guanine-nucleotide exchange factors. Gαq is inactivated by regulators of G protein signaling proteins, as well as by PLC-β3. Gαq further interacts with G protein-coupled receptor kinase 2 (GRK2), although the functional role of this interaction is debated.
Here, we map the structural determinants of Gαq interactions with multiple partners
(i.e., Gαq multi-specificity) using structure-based energy calculations. We delineate regions that specifically interact with GTPase Activating Proteins (GAPs) and residues that exclusively contribute to effector interactions, showing that only the Gαq "Switch II" region interacts with all partners. Our analysis further suggests that Gαq-GRK2 interactions are consistent with GRK2 functioning as an effector, rather than a GAP. Our multi-specificity analysis pinpoints Gαq residues that uniquely contribute to interactions with particular partners, enabling precise manipulation of these cascades. As such, we dissect the molecular basis of Gαq function as a central signaling hub, which can be used to target Gαq-mediated signaling in therapeutic interventions.
|
|
 |
|
Residue-level determinants of angiopoietin-2 interactions with its receptor Tie2.
Anna Bakhman, Eitan Rabinovich, Tomer Shlamkovich, Niv Papo and Mickey Kosloff
Proteins: Structure, function and Bioinformatics. (2019) 87 (3): 185-197.
web
pdf (3 Mb)
In this work we dissect the protein-protein interactions of angiopoietin2 (Ang2) with its Receptor
Tyrosine Kinase (RTK) Tie2. These interactions have broad biological importance, as they regulate
physiological processes such as angiogenesis and vascular permeability. Ang2 binding to Tie2 also
plays a central role in pathologies such as inflammation, autoimmune diseases, sepsis, ophthalmic
diseases, developmental abnormalities, and tumorigenesis - making this system an attractive drug
target. Relevantly, the comparable VEGF system is already a validated target for widely used drugs,
but broad gaps persist in understanding Ang2 biology.
We used physics-based electrostatic and surface-area calculations to identify the subset of
interfacial Ang2 and Tie2 residues that can affect binding directly. Using random and site-directed
mutagenesis and yeast surface display (YSD), we validated these predictions and identified additional
Ang2 positions that affected receptor binding. We then applied a new method we developed here that
uses burial-based calculations to classify the larger set of Ang2 residues that are buried in the Ang2
core. Mutations in such residues can perturb the Ang2 structure and thereby affect interactions with
Tie2 indirectly. Our analysis showed that the Ang2-Tie2 interface is dominated by nonpolar
contributions, with only three Ang2 and two Tie2 residues that contribute electrostatically to
intermolecular interactions. Individual interfacial residues contributed only moderately to binding,
suggesting that engineering of this interface will require multiple mutations to reach major effects.
Conversely, substitutions in substantially buried Ang2 residues were more prevalent in our
experimental screen, reduced binding substantially, and are therefore more likely to have a
deleterious effect that might contribute to oncogenesis. Computational analysis of additional
RTK-ligand complexes, c-Kit/SCF and M-CSF/c-FMS, and comparison to previous YSD results, further show
the utility of our combined methodology.
|
|
 |
|
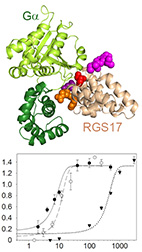
Structural motifs in the RGS RZ subfamily combine to attenuate interactions with Gα
subunits.
Denise Salem-Mansour, Ali Asli, Meirav Avital-Shacham and Mickey Kosloff
Biochem. Biophys. Res. Commun. (2018) 503 (4): 2736-2741.
web
pdf (3.6 Mb)
The RGS RZ subfamily, which consists of RGS17, RGS19, and RGS20, mediates numerous physiological
functions and human pathologies - mostly by functioning as GTPase Activating Proteins (GAPs) towards
the Gα subfamily. Yet, which RZ subfamily members mediate particular functions and how their GAP
activity and specificity are governed at the amino acid level is not well understood.
Here, we show using biochemical assays that all RZ subfamily members have similar and relatively low
GAP activity towards Gαo. We characterized four RZ-specific structural motifs that mediate this
low
activity, and suggest they perturb optimal interactions with the Gα subunit. Indeed, inserting
these
RZ-specific motifs into the representative high-activity RGS16 impaired GAP activity in a non-additive
manner. Our results provide residue-level insights into the specificity determinants of the RZ
subfamily, and enable to study their interactions in signaling cascades by using the redesigned
mutants that we tested in this work.
|
|
 |
|
Neofunctionalization in Ligand Binding Sites of Ant Olfactory Receptors.
Rana Saad, Amir Cohanim, Mickey Kosloff and Eyal Privman
Genome Biol. Evol. (2018) 1 (10): 2490-2500.
web
pdf (3 Mb)
Insect olfactory receptors (ORs) are the largest family of chemosensory receptors. Ants are an extreme
example with ~400 ORs per genome - the highest number in insects - presumably reflecting an increased
complexity of chemical communication. Here, we examined gene duplications and positive selection on
ant ORs, reconstructing the hymenopteran OR gene tree and inferring positive selection along every
branch using the branch-site test. We find more positive selection in branches following
species-specific duplications and, using 3D structural homology, identified amino acid sites targeted
by positive selection and mapped them onto a structural model of insect ORs. The latter form two
clusters on the extracellular side of the receptor, on either side of a cleft in the structure. This
region was previously implicated in ligand activation, suggesting that the concentration of positively
selected sites in this region is related to adaptive evolution of ligand binding sites or allosteric
transmission of ligand activation. These results provide insights into chemical signaling in ant
societies and the specific OR subfamilies and individual residues that facilitated adaptive evolution
of olfactory functions.
|
|
 |
|
Interplay between negative and positive design elements in Gα helical domains of G proteins
determines interaction specificity toward RGS2.
Mohammad Kasom, Samia Gharra, Isra Sadiya, Meirav Avital-Shacham and Mickey Kosloff
Biochem. J. (2018) 475 (14): 2293-2304.
web
pdf (3.9 Mb)
Among all RGS proteins, RGS2 is unique in interacting only with the Gαq but not with the
Gα subfamily.
Previous studies suggested that this specificity is determined by the RGS domain and, in particular,
by three RGS2-specific residues that lead to a unique mode of interaction with Gαq. This
interaction
was further proposed to act through contacts with the Gα GTPase domain.
Here, we combined energy calculations and GTPase activity measurements to determine which Gα
residues
dictate specificity toward RGS2. We identified putative specificity-determining residues in the
Gα
helical domain, which among G proteins is found only in Gα subunits. Replacing these helical
domain
residues in Gα with their Gαq counterparts resulted in a dramatic specificity switch
toward RGS2. We
further show that Gα-RGS2 specificity is set by Gα residues that perturb interactions with
RGS2, and
by Gαq residues that enhance these interactions. These results show, for the first time, that
the Gα
helical domain is central to dictating specificity toward RGS2, suggesting that this domain plays a
general role in governing Gα-RGS specificity.
|
|
 |
|
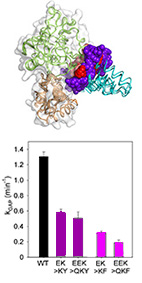
"Disruptor" residues in the regulator of G protein signaling (RGS) R12 subfamily attenuate
the inactivation of Gα subunits.
Ali Asli, Isra Sadiya, Meirav Avital-Shacham and Mickey Kosloff.
Science Signal. (2018) 11 (534): aan3677
web
pdf (1.6 Mb)
The RGS R12 subfamily, which consists of RGS10, RGS12, and RGS14, mediates numerous physiological
functions and human pathologies - mostly by functioning as negative regulators of Gα subunits.
However, the molecular and structural mechanism that enable specific interactions between these two
large families are not well understood.
Here, we used biochemical assays to show that R12 subfamily members RGS10 and RGS14 have lower
activity than most RGS R4 subfamily members toward the subfamily member Gαo. Using
structure-based
energy calculations with multiple Gα-RGS complexes, we identified R12-specific residues in
positions
that are predicted to determine the divergent activity of this subfamily. This analysis predicted that
these residues, which we call "disruptor residues", interact with the Gα helical
domain. We
engineered the R12 disruptor residues we discovered into the RGS domains of high-activity R4 subfamily
members and found that these altered proteins exhibited reduced activity toward Gαo.
Reciprocally,
replacing the putative disruptor residues in RGS18 (a member of the R4 subfamily that also exhibited
low activity toward Gαo) with the corresponding residues from a high-activity R4 subfamily RGS
protein
increased its activity toward Gαo. Furthermore, the high activity of the R4 subfamily toward
Gαo was
independent of the residues in the homologous positions to the R12 subfamily and RGS18 disruptor
residues. Thus, our results suggest that the identified RGS disruptor residues function as negative
design elements that attenuate RGS activity for specific Gα proteins, and define a new
specificity-determining type of motif.
|
|
 |
|
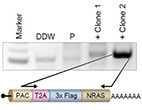
Refinement of the endogenous epitope tagging technology allows the identification of a novel NRAS
binding partner in melanoma.
Michal Alon, Rafi Emmanuel, Nouar Qutob, Anna Bakhman, Victoria Peshti, Alexandra Brodezki, David
Bassan, Mickey Kosloff and Yardena Samuels
Pigment Cell Melanoma Res. (2018) 31 (5): 641-648.
web
pdf (1.2 Mb)
The NRAS oncoprotein is highly mutated in melanoma. Here, we adapted the endogenous epitope tagging (EET) approach and utilized it to identify novel NRAS binding partners. Using EET, an epitope tag is added to the endogenously expressed protein via modification of its genomic coding sequence. Existing EET systems are not robust, suffer from high background, and are labor-intensive.
Here, we developed a polyadenylation signal-trap construct for N'-tagging that generates a polycistronic mRNA with the gene of interest, integrating the tagging cassette in frame with the
target gene. Using this design, we demonstrated for the first time endogenous tagging
of NRAS in melanoma cells. Thereby, we identified the E3 ubiquitin ligase c-CBL as a novel NRAS
binding partner. Using an extensive structural comparison of different RAS isoforms and mutants, we
showed they are essentially identical in structure and suggested they bind these particular protein
partners similarly. The EET technology we developed allows the characterization of new RAS effectors, which could be beneficial for the design of future drugs that inhibit constitutive signaling of RAS oncogenic mutants.
|
|
 |
|
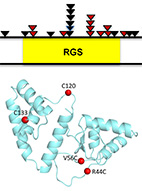
RGS7 is recurrently mutated in melanoma and promotes migration and invasion of human cancer
cells.
Nouar Qutob, Ikuo Masuho, Michal Alon, Rafi Emmanuel, Isadora Cohen,
Antonella Di Pizio, Jason Madore, Abdel Elkahloun, Tamar Ziv, Ronen Levy,
Jared Gartner, Victoria Hill, Jimmy Lin, Yael Hevroni, Polina Greenberg,
Alexandra Brodezki, Steven Rosenberg, Mickey Kosloff, Nicholas Hayward,
Arie Admon, Masha Niv, Richard Scolyer, Kirill Martemyanov and Yardena Samuels
Sci. Rep. (2018) 8 (1): 653.
web
pdf (1.3 Mb)
Sup. Materials (2.3 Mb)
Many studies have linked various RGS proteins to cancer, yet few molecular details are know on how
RGS proteins might be involved in tumorigenesis. Here, we analyzed 501 melanoma exomes to reveal that
RGS7
is a tumor-suppressor gene that was mutated in 11% of melanomas. In particular, RGS7 was found to
harbor
three recurrent mutations (p.R44C, p.E383K and p.R416Q).
Structural modeling of the most common of these (p.R44C)
predicted that it destabilizes the protein due to the loss of an electrostatic/H-bond network.
We confirmed this prediction experimentally, showing that the R44C mutant is indeed destabilized
and has weaker catalytic activity towards Gαo, thus providing a dual mechanism for its loss of
function.
Both of these effects can contribute to RGS7 loss of function,
resulting in increased anchorage-independent growth, migration and invasion of melanoma cells.
We further identified an additional mutation that restored catalytic activity and reinstated protein
stability - leading to the rescue of RGS7 function as a tumor suppressor.
Our findings identify RGS7 as a novel melanoma driver and point to the clinical
relevance of using strategies to stabilize the protein and, thereby, restore its function.
|
|
 |
|
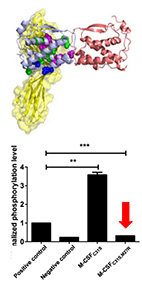
Engineering a monomeric variant of macrophage colony-stimulating factor
(M-CSF) that antagonizes the c-FMS receptor.
Yuval Zur, Lior Rosenfeld, Anna Bakhman, Stefan Ilic, Hezi Hayun,
Anat Shahar, Barak Akabayov, Mickey Kosloff, Noam Levaot and Niv Papo
Biochem. J. (2017) 474 (15): 2601-2617.
web
pdf (1.5 Mb)
Dysregulation and overexpression of macrophage colony-stimulating factor (M-CSF)
and its c-FMS tyrosine kinase receptor, proteins that are essential for osteoclast differentiation,
are known to promote bone metastasis and osteoporosis, making both the ligand and its
receptor attractive targets for therapeutic intervention.
With this aim in mind, our starting point was the previously held concept that the potential
of the M-CSF C31S mutant as a therapeutic is derived from its inability to dimerize and hence
to act as an agonist.
However, here we showed that dimerization is not abolished in the C31S mutant as expected and
that it retains agonistic activity toward osteoclasts. We solved the crystal structure of the C31S
dimer complex and used structure-based energy calculations to identify the residues responsible
for its dimeric form. We used this analysis to develop a double M-CSF mutant and showed that is
functions as a high-affinity antagonist for c-FMS -retaining its binding ability but preventing
ligand dimerization. The monomeric properties of this mutant were validated using dynamic light
scattering and small-angle X-ray scattering analyses. We also showed that this mutant is a functional
inhibitor of M-CSF-dependent c-FMS activation and osteoclast differentiation in vitro.
Our study, therefore, provides insights into the sequence-structure-function relationships
of M-CSF/c-FMS interactions and of ligand/receptor tyrosine kinase interactions in general.
|
|
 |
|
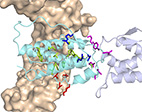
Identifying Residues that Determine SCF Molecular-Level Interactions
through a Combination of Experimental and In silico
Analyses.
Eitan Rabinovich, Michael Heyne, Anna Bakhman, Mickey Kosloff,
Julia Shifman and Niv Papo
J. Mol. Biol. (2017) 429 (1): 97-11.
web
pdf (2.3 Mb)
The complex of stem cell factor (SCF) with its partner, the receptor
tyrosine kinase c-Kit, plays significant roles in hematopoiesis and
angiogenesis and is an attractive target for rational drug design. Yet,
the residue-level details of the SCF/c-Kit interaction sites and the
mechanisms that modulate this interaction are not well understood.
Here, we use protein-wide epitope mapping using yeast surface display
(YSD) to interrogate these interactions. Using a library of single SCF
mutants that span the SCF sequence we identified variants with decreased
affinity to c-Kit. Screening of these SCF clones for binding to a
structural antibody helped identify mutations that result in small or
large conformational changes in SCF. Using computational modeling we
showed that these mutations reduced the binding affinity through one of
the three mechanisms: through SCF destabilization, through elimination of
favorable SCF/c-Kit intermolecular interactions, or through allosteric
changes. Experimentally measured in vitro binding affinities of purified
mutants confirmed both the yeast surface display selection results and
the computational predictions. Our study thus identified the residues
crucial for c-Kit/SCF binding and demonstrated the advantages of using a
combination of computational and combinatorial methods for RTK-ligand
epitope mapping.
|
|
 |
|
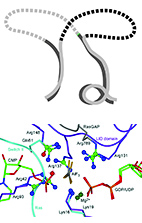
From Protein Structure to Function via Computational Tools and Approaches.
Rachel Kolodny and Mickey Kosloff
Isr. J. Chem. (2013) 53 (3-4):147-156.
web
pdf (1.5 Mb)
The 3D structures of proteins are often considered fundamental for
understanding their function.
Yet, because of the complexity of protein structure,
extracting specific functional information from structures can be a
considerable challenge.
Here, we present selected approaches and tools that were developed in
the Kolodny and Kosloff labs to study and connect protein sequence,
structure, and function spaces.
First, we consider a global perspective of structure space and
view the protein data bank (PDB) as a database.
We highlight challenges in searching protein structure space and
in using the PDB as the starting point for computational structural studies.
Then we describe a function-oriented view and show examples of how
multiple protein structures can be used to extract insights about the
function and specificity of proteins at the family level.
|
|
|
|
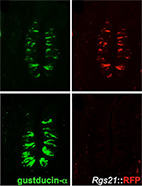
Regulator of G-protein signaling-21 (RGS21) is an inhibitor of bitter gustatory signaling found in
lingual and airway epithelia.
Staci Cohen, Brian Buckley, Mickey Kosloff, Alaina Garland, Dustin Bosch, Gang Cheng Jr, Harish
Radhakrishna, Michael Brown, Francis Willard, Vadim Arshavsky, Robert Tarran, David Siderovski and
Adam Kimple
J. Biol. Chem. (2012) 287 (50): 41706-41719.
web
pdf (4.7 Mb)
RGS21 is a newly discovered RGS protein, whose biological function and specificity are unclear.
The recent cloning of RGS21 from taste bud cells has implicated this protein in the regulation
of taste signaling - presumably via the Gi family members gustducin and transducin (Gt),
which have been implicated in these sensory cascades.
However, the exact role of RGS21 has not been precisely defined.
Here, we sought to determine the role of RGS21 in tastant responsiveness.
Our computational analysis predicted that RGS21 is a highly active and promiscuous GAP toward
members of the Gi subfamily.
Indeed, our biochemical experiments confirmed that RGS21 shows high GAP activity towards Gt and Go.
We further demonstrated that RGS21 is not only endogenously expressed in mouse taste buds but
also in lung airway epithelial cells, which have previously been shown to express components
of the taste signaling cascade.
Furthermore, the immortalized human airway cell line 16HBE was found to express transcripts
for tastant receptors, RGS21, and downstream taste signaling components.
Over- and underexpression of RGS21 in 16HBE cells confirmed that RGS21 acts to oppose
bitter tastant signaling to cAMP and calcium second messenger changes.
Our data collectively suggests that RGS21 modulates bitter taste signal transduction.
|
|
|
|
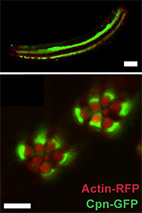
Compartmentalization and Ca2+ buffering are essential for prevention of light-induced retinal
degeneration.
Shirley Weiss, Elkana Kohn, Daniela Dadon, Ben Katz, Maximilian Peters, Mario Lebendiker, Mickey
Kosloff, Nansi Jo Colley and Baruch Minke
J. Neurosci. (2012) 32 (42): 14696-14708.
web
pdf (3.6 Mb)
Fly photoreceptors are polarized cells, each of which has an extended interface between its cell
body and the light-signaling compartment, the rhabdomere.
Upon intense illumination, rhabdomeric calcium concentration reaches millimolar levels that would
be toxic if calcium diffusion between the rhabdomere and cell body was not robustly attenuated.
Yet, it is not clear how such effective attenuation is obtained.
Here, we show that calcium homeostasis in the photoreceptor cell relies on the protein calphotin,
which has a unique amino acid composition and is predicted to be intrinsically-unstructured.
This unusual protein functions as an immobile calcium buffer localized along the base of the
rhabdomere,
separating the signaling compartment from the cell body.
Generation and analyses of transgenic Drosophila strains, in which calphotin-expression levels were
reduced in a graded manner, showed that moderately reduced calphotin expression impaired calcium
homeostasis while calphotin elimination resulted in severe light-dependent photoreceptor degeneration.
The degeneration was rescued by prevention of calcium overload via overexpression of CalX,
the Na(+)-Ca(2+) exchanger.
In addition, reduced calphotin levels resulted in abnormally fast kinetics of calcium elevation
in photoreceptor cells.
Together, our data suggest that calphotin functions as a calcium buffer.
We propose that calphotin-mediated compartmentalization and calcium buffering constitute an effective
strategy to protect cells from calcium overload and light-induced degeneration.
Further analysis of Calphotin's sequence and amino acid composition raises the intriguing possibility
that calphotin is a prototypic example of a new and diverse family of intrinsically-unstructured
calcium-binding proteins in diverse organisms.
|
|
 |
|
Cooperative phosphoinositide and peptide binding by PSD-95/discs large/ZO-1 (PDZ) domain of
polychaetoid, Drosophila zonulin.
Ylva Ivarsson, Anna Maria Wawrzyniak, Gunther Wuytens, Mickey Kosloff, Elke Vermeiren, Marie Raport
and Pascale Zimmermann
J. Biol. Chem. (2011) 286 (52): 44669-44678.
web
pdf (3.3 Mb)
PDZ domains are well known protein-protein interaction modules that assemble molecular complexes as
part of multidomain proteins. Some PDZ domains have been reported to interact with membrane lipids
(particularly phosphoinositides), but few studies have elucidated the molecular details of such
interactions.
Here, we screened 46 Drosophila PDZ domains for phosphoinositide-dependent cellular localization and
discovered that the second PDZ domain of polychaetoid (Pyd PDZ2) interacts with phosphatidylinositol
4,5-bisphosphate at the plasma membrane. Surface plasmon resonance binding experiments established
that Pyd PDZ2 interacts with phosphoinositides with micromolar range affinity. We then analyzed a
homology model of dimeric Pyd PDZ2 and showed that electrostatic interactions involving an extended
positively charged surface of Pyd PDZ2 are crucial for its phosphoinositide-dependent membrane
interactions. We then validated these predictions by mutagenesis studies followed by binding and
localization experiments. Furthermore, we show that both lipid and peptide binding contribute to the
membrane localization of Pyd PDZ2, raising the intriguing possibility that this PDZ domain may
coordinate protein- and phospholipid-mediated signals.
|
|

|
|
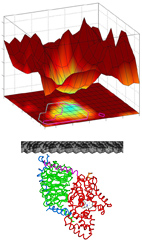
Integrating energy calculations with functional assays to decipher
the specificity of G protein-RGS protein interactions.
Mickey Kosloff, Amanda Travis, Dustin Bosch, David Siderovski and Vadim Arshavsky
Nature Struct. Mol. Biol. (2011) 18 (7): 846-853.
web
pdf (2.3 Mb)
Sup. Materials (600 Kb)
External coverage:
F1000 evaluation
HFSP
coverage
We describe an approach to address a fundamental challenge in the field of signal
transduction - deciphering how protein structure encodes specific interactions between
large protein families. As a model system to study specificity,
we chose to investigate the interactions of heterotrimeric G-proteins with
Regulators of G-protein Signaling (RGSs).
RGSs are responsible for turning G-proteins "off" and in multiple cascades across
most human tissues RGSs determine the duration of the G-protein coupled signaling.
RGSs have also been implicated in a wide range of human pathologies and are
promising drug targets, both as primary targets and as complements to drugs
that target other components in G-protein signaling (such as G-protein Coupled
Receptors).
To understand how RGS protein structure encodes both their common ability to inactivate
G-proteins but also their selective G-protein recognition, we integrated
structure-based energy calculations with biochemical measurements of RGS protein
activity. Using a consensus approach across the eight available RGS-domain/G-protein
crystal structures, we established a structure-to-sequence map predicting which RGS
residues are essential for function and which RGS residues can modulate specific
interactions with the cognate G-protein. This map revealed that, in addition to
previously identified conserved residues, RGS proteins contain another group of
variable "Modulatory Residues", which reside at the periphery of the
RGS-domain/G-protein interface and fine-tune G-protein recognition. Mutations of
Modulatory Residues in high-activity RGS proteins impaired RGS function, whereas
redesign of low-activity RGS proteins in critical Modulatory positions yielded complete
gain-of-function RGS mutants. Therefore, RGS proteins combine a conserved core
interface with peripheral Modulatory Residues to selectively optimize G-protein
recognition and inactivation.
Finally, we applied this computational approach to a completely different system - the
interactions of colicin E7 with its inhibitory immunity proteins, a well-established
model for studying protein-protein interaction specificity. We thereby showed the
generality of our structure-based method and its potential use in a scalable
"bottom-up" approach to study the structural basis for the "wiring" of signal
transduction networks.
|
|
 |
|
Electrostatic and Lipid-Anchor Contributions to the Interaction of Transducin with Membranes:
Mechanistic Implications for Activation and Translocation.
Mickey Kosloff, Emil Alexov, Vadim Arshavsky and Barry Honig
J. Biol. Chem. (2008) 283 (45): 31197-31207.
web
pdf (900 Kb)
Sup. Materials (1.3 Mb)
The heterotrimeric G-protein transducin is a key component of the vertebrate
phototransduction cascade. Like many other signal transduction proteins,
transducin is peripherally attached to membranes of the rod outer segment,
where it interacts with other proteins at the membrane-cytosol interface.
Here, we used a computational approach to analyze the interaction strength
of transducin and its subunits with membranes, as well as the range of orientations
that they are allowed to occupy on the membrane surface. We show that the
membrane-bound transducin heterotrimer is constrained to a limited range of
orientations, which can accelerate transducin's activation by rhodopsin.
Notably, the membrane interactions of the dissociated transducin subunits are very
different from those of the heterotrimer. While the beta-gamma complex is attracted to
the negatively charged membrane, we show that Gt-alpha is electrostatically repelled by
such membranes.
We suggest that this repulsion could facilitate the membrane dissociation and
intracellular translocation of Gt-alpha. Moreover, we show that the properties
described for transducin are common to its homologs within the Gi subfamily.
In a broader view, this work exemplifies how the activity-dependent association
and dissociation of a G-protein can change both the affinity for membranes and the
range of allowed orientations, thereby modulating G-protein function.
Importantly, our approach can characterize quantitatively the interactions of
other peripheral membrane proteins with membranes.
|
|
 |
|
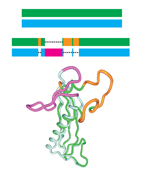
Sequence-Similar, Structure-Dissimilar Proteins in the PDB.
Mickey Kosloff and Rachel Kolodny
Proteins: Structure, function and Bioinformatics. (2008) 71 (2):
891-902.
web
pdf (500 Kb)
Sup. Materials (250 kb)
It is often assumed that in the Protein Data Bank (PDB),
two proteins with similar sequences will also have similar structures.
This assumption underlies many computational studies and structure prediction methods.
Here, we compare sequence-based structural superpositions and geometry-based
structural alignments and show that the former provides
a better measure of structure dissimilarity.
Using sequence-based structural superpositioning we find many examples in the PDB where
two proteins that are similar in sequence have structures that differ significantly
from one another, usually in direct relation to
their function. We conclude that the assumption of two proteins with similar
sequences having similar structures is often incorrect
and can lead to the loss of structurally and functionally important information.
We have established a
database of sequence-similar, structurally dissimilar protein pairs
that will help address this problem and show how this database can assist in predicting structures
using
homology modeling.
|
|
 |
|
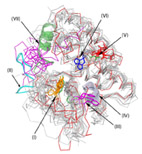
Comparative Structural Analysis of a Novel Glutathoine S-transferase (Atu5508) from
Agrobacterium tumefaciens at 2.0 Angstrom Resolution.
Mickey Kosloff et al.
(with JCSG consortium)
Proteins: Structure, function and Bioinformatics. (2006) 65 (3): 527-537.
web
pdf (1 Mb)
Glutathione S-transferases (GSTs) comprise a diverse superfamily of enzymes found in organisms from
all kingdoms of life.
They are involved in diverse processes, notably small-molecule biosynthesis and detoxification,
and are frequently used in protein engineering studies and as biotechnology tools.
Because the GST superfamily is very diverse, GSTs have been subdivided into an ever-increasing number
of sub-families, or “classes”, associated with different functionalities and enzymatic
specificities.
This classification has usually been based on a combination of criteria, such as biochemical
properties, primary,
tertiary and quaternary structure and immunological reactivity.
Here, through use of comparative sequence and structural analysis of the GST superfamily,
we identified local sequence and structural signatures that allowed us to distinguish between
different GST classes.
Uniquely, this approach enables classifying novel GST proteins based on structure only,
without requiring additional biochemical or immunological data.
In this work we also report the high-resolution X-ray structure of Atu5508, a putative GST from the
pathogenic soil bacterium
Agrobacterium tumefaciens (atGST1, PDB id 2FNO).
Our comparative structural analysis suggests that atGST1 defines a new GST class,
distinct from previously characterized GSTs both in structure and in function,
which makes it an attractive target for further biochemical studies.
Importantly, our comparative analysis and characterization of atGST1 were first performed without any
knowledge of the
experimental structure, when we were “blindly“ predicting atGST1's 3D structure during
CASP6. At the time, all available GST structures
(templates)
had less than 20% sequence identity to atGST1, yet we were able to successfully predict both the 3D
structure and its functionality.
Later, our approach and conclusions were corroborated using the experimental structure, thus
validating the use of this approach to predict
active site specificity.
|
|
 |
|
GTPase Catalysis by Ras and Other G-proteins: Insights from Substrate Directed
SuperImposition.
Mickey Kosloff and Zvi Selinger
J. Mol. Biol. (2003) 331 (5): 1157-1170.
web
pdf (2.5 Mb)
Comparisons of different protein structures are commonly carried out by
superimposing the coordinates of the protein backbones or
selected parts of the proteins. However, when the objective is analysis of
similarities and differences in enzyme active sites,
there is an inherent problem in using the domains under investigation for the superimposition.
In this work we use a comparative approach we termed “Substrate Directed SuperImposition”
(SDSI).
It entails the superimposition of multiple protein-substrate structures
using exclusively the coordinates of the comparable substrates.
SDSI has the advantage of unbiased comparison of the active-site
environment from the substrate’s point of view.
Here we apply SDSI to various G-protein structures for dissecting the mechanism of the
GTPase reaction. SDSI indicates that dissimilar G-proteins stabilize the transition state
of the GTPase reaction similarly and supports the commonality of the critical step in GTPase -
the reorientation of the critical arginine and glutamine. We ascribe the catalytic inefficiency
of the small G-protein Ras to the great flexibility of its active site and downplay possible
catalytic roles for the Lys16 residue in GTPase catalysis.
We also show that in contrast to all other Gly12 Ras mutants, which are oncogenic,
the Gly12->Pro mutant does not interfere with the catalytic orientation of the critical glutamine.
This suggests why this mutant has a higher rate of GTP hydrolysis and is non-transforming.
Finally, we use SDSI to compare enzymes with very different 3D structures to reveal surprising
similarities in
the divergent catalytic machineries of G-proteins and UMP/CMP kinases.
|
|
 |
|
Regulation of Light-dependent Gq-alpha Translocation and Morphological Changes in Fly
Photoreceptors
Mickey Kosloff, Natalie Elia, Tamar Joel-Almagor, Rina Timberg, Troy Zars, David Hyde, Baruch Minke
and Zvi Selinger
EMBO J. (2003) 22 (3): 459-468.
web1
web2
pdf (400 Kb)
Heterotrimeric G-proteins relay signals between membrane-bound G-protein coupled receptors (GPCRs) and
downstream effectors.
To perform their signaling function, G-proteins require anchorage to the plasma membrane.
While in vitro and cell line based investigations suggested that the membrane localization of
G-proteins is reversible,
the results are contradictory. Importantly, these phenomena have not been characterized in vivo and
the regulation of
G-alpha localization within the natural endogenous environment of a specialized signaling cell is
therefore
of great interest.
Here we show, using live Drosophila flies, that light causes massive and reversible translocation of
the visual Gq-alpha subunit
from the membrane to the cytosol. This translocation is associated with marked architectural changes
in the signaling compartment.
We characterize the translocation cycle and how signaling molecules that interact with Gq-alpha
regulate these processes.
We also show that Gq-alpha is necessary but not sufficient to bring about the morphological changes in
the signaling organelle.
Furthermore, mutant analysis indicates that Gq-beta is essential for targeting of Gq-alpha to the
membrane and suggests that
Gq-beta is also needed for efficient activation of Gq-alpha by rhodopsin.
|
|
 |
|
Structural Homology Discloses a Bifunctional Structural Motif at the N-termini of G alpha
Proteins.
Mickey Kosloff, Natalie Elia and Zvi Selinger
Biochemistry (2002) 41 (49): 14518-14523.
web
pdf (800 Kb)
Lipid modification by palmitoylation is a fundamental contributor to the
membrane localization of heterotrimeric G-proteins
and other proteins, but the signals leading to this reversible modification are still unknown.
Here, we use homology models of different human G-alpha paralogs (generated with automated methods) to
identify a basic,
positively charged structural motif in the N-termini of these proteins.
We also show that a similar structural motif is found in other palmitoylated proteins.
This basic motif is not readily discernible from sequence alone and is found in all palmitoylated-only
G-proteins.
Contrastingly, G-alpha subunits that also undergo myristoylation do not contain these prominent basic
patches,
suggesting that this basic motif and myristoylation play overlapping roles in membrane targeting.
|
|

|
|
Substrate Assisted Catalysis - Application to G-proteins.
Mickey Kosloff and Zvi Selinger
TiBS (2001) 26 (3): 161-168.
web
pdf (720 Kb)
In Substrate-Assisted Catalysis (SAC) the substrate for enzymatic catalysis provides one or more
functional groups that actively
participate in the catalytic process.
Here, we describe how SAC is applicable to guanine nucleotide-binding proteins (G proteins) in two
different aspects:
1) naturally occurring SAC uses GTP as a general base in the GTPase reaction catalyzed by G proteins.
2) Engineered SAC has identified a putative rate-limiting step for the GTPase reaction and shown that
GTPase-deficient
oncogenic Ras mutants are not irreversibly impaired.
We use a novel structure superimposition approach to analyze the anatomy of different G-proteins'
active sites and the role of
specific residues in the GTPase mechanism. We also discuss why the role of G-proteins as molecular
switches evolved catalytic inefficiency.
We describe the implications for the catalytic mechanism and how SAC paves the way to designing novel
anti-cancer drugs
to restore the blocked GTPase reaction that drives many human cancers.
|
|
 |
|
Substrate-Assisted Catalysis: Implications for Biotechnology and Drug Design.
Mickey Kosloff, Tsaffrir Zor and Zvi Selinger
Drug Dev. Res. (2000) 50: 250-257.
web
pdf (135 Kb)
In Substrate-Assisted Catalysis (SAC) the substrate for enzymatic catalysis provides one or more
functional groups that
actively participate in the catalytic process.
Here, we describe the occurrence of SAC in natural enzymes and its use as an engineered tool to study
catalytic mechanisms.
We explain how this paves the way to novel therapeutic and biotechnological approaches aimed at
restoring the activity of mutant
inactive enzymes.
|
|
| |
|

